
科研进展
National Science Review |康岚/魏珂/高绍荣团队揭示丙酮酸激酶PKM1在胚胎心肌细胞增殖中的关键作用
发布时间:2025-09-30
心脏是哺乳动物胚胎期最早发挥功能的器官,其发育过程中既要维持强大的能量供给以保证搏动,又要通过心肌细胞的增殖不断扩大体积和完善结构,从而为出生后的循环系统功能奠定基础。糖酵解关键酶丙酮酸激酶M型(PKM)存在两种剪接亚型,PKM1和PKM2,在能量供应和代谢平衡中发挥不同作用。PKM1以高活性的形式广泛存在于高能耗组织中,然而此前由于PKM1敲除常常伴随PKM2代偿性上调,其在心脏发育中的独立作用一直存在争议。阐明PKM1在胚胎心肌细胞增殖及心脏发育中的真实功能,对于理解心脏能量代谢与结构发育之间的联系具有重要意义。
近日,同济大学生命科学与技术学院康岚教授、高绍荣院士团队与魏珂教授团队在National Science Review期刊上在线发表了题为“PKM1 is required for embryonic cardiomyocyte proliferation through energetic regulation of NFYa stability”的研究论文。该工作通过构建一种在PKM1缺失背景下不伴随PKM2代偿性上调的小鼠模型,系统揭示了PKM1在维持能量稳态和促进心肌细胞增殖中的关键作用。

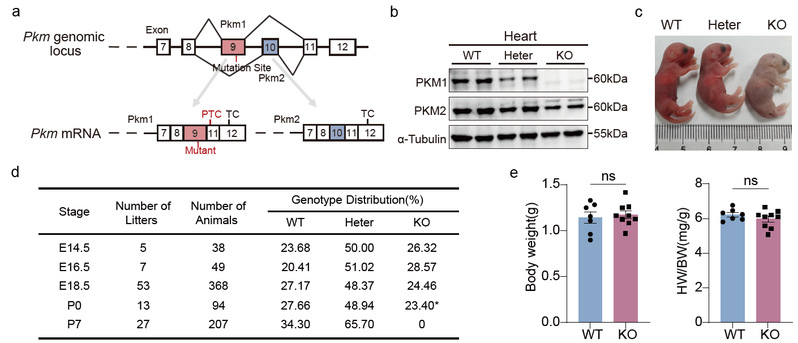
研究团队首先在 Pkm1 特异性外显子引入突变,成功建立了 Pkm1 缺失小鼠模型。与传统的条件性敲除不同,这一策略有效避免了 PKM2 的上调,从而真实反映 PKM1 的缺失效应。结果发现,虽然杂合子小鼠可以正常存活,但PKM1完全缺失的小鼠在出生后数小时内全部死亡,表现出包括心室致密层变薄、房间隔缺损以及心肌收缩功能受损的心脏结构和功能异常。缺失PKM1的心肌细胞增殖能力显著下降,而细胞凋亡并未增加,同时伴随细胞肥大现象,提示致密层发育不良主要由增殖受损引起。该表型自胚胎发育晚期(约E16.5)开始显现,与PKM1在胚胎晚期心脏中表达上调的时相一致。
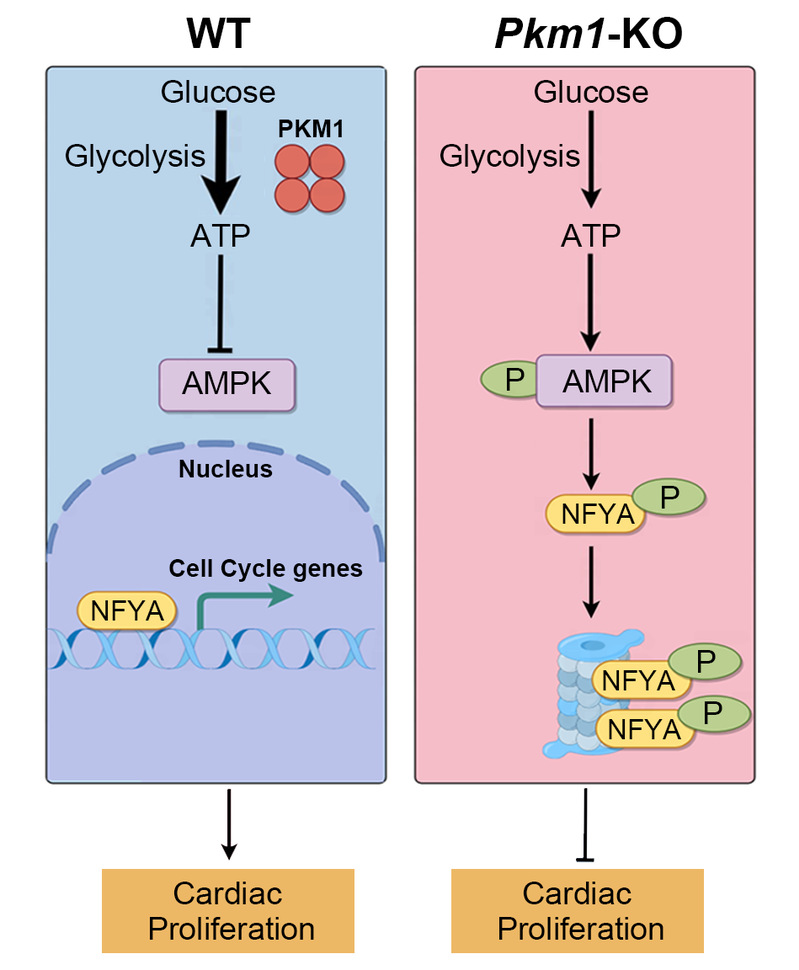
机制研究表明,PKM1缺失导致糖酵解阻滞并引起酸烯醇式丙酮酸累积,心肌组织ATP水平下降,线粒体呼吸和膜电位减弱,能量供应受到严重破坏。能量不足触发了能量感受器AMPK的持续激活,而AMPK在心肌细胞中过度活化会显著抑制增殖。通过药理学干预发现,抑制AMPK活性能够在一定程度上恢复PKM1缺失或敲低条件下的心肌细胞增殖能力,提示AMPK是心肌代谢失衡与增殖缺陷之间的关键枢纽。
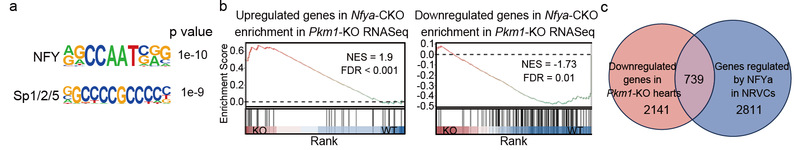
转录组学和功能分析进一步锁定转录因子NFYa作为下游重要效应分子。PKM1缺失并未影响Nfya转录水平,但NFYa蛋白水平显著下降,原因在于激活的AMPK使NFYa蛋白第325位丝氨酸磷酸化,继而导致该蛋白的降解。功能性实验表明,无论在小鼠心肌还是在人源诱导多能干细胞来源的心肌细胞中,NFYa过表达均能部分恢复因PKM1缺失导致的增殖受损,验证了“PKM1-AMPK-NFYa”轴在心脏发育和心肌增殖调控中的保守性和关键作用。
综上,该研究首次在排除PKM2代偿性上调的背景下,明确了PKM1在胚胎心肌细胞增殖和心脏发育中的必需作用,提出了一个从代谢稳态、能量感知到转录调控的分子机制框架。研究揭示PKM1通过维持能量供给抑制AMPK异常激活,从而保护NFYa稳定性,进而驱动心肌细胞增殖和心脏结构的正常形成。这一发现不仅丰富了我们对能量代谢与心脏发育之间关系的理解,也为阐释先天性心脏病的代谢基础及未来潜在干预策略提供了新的理论依据。
同济大学康岚教授、魏珂教授、高绍荣院士为论文的共同通讯作者。同济大学博士研究生张丹丹、博士后唐岩松、博士研究生叶文为论文的共同第一作者。该研究得到了国家重点研发计划和国家自然科学基金等项目的重要支持。
Copyright© 2011-2015 生命科学与技术学院, All rights reserved
地址:上海市四平路1239号 电话:021-65981041 传真:65981041
