
科研进展
Nucleic Acids Research|史偈君/刘君等开发核酸修饰测序通用比对算法
发布时间:2024-12-17

随着表观遗传学和表观转录组学的快速发展,近年来涌现出大量RNA修饰和DNA修饰的检测方法。其中碱基转换(Base Conversion,简称为BC)方法由于能达到单碱基分辨率,对下游靶标发现和机制探索最为有利。按照碱基转换方式,BC 方法可分为三类:单路转换,如用于检测 5mC 的 C-to-T 转换,或用于检测 m6A 的 A-to-G 转换【1,2】;多路转换,如用于检测 m1A 的A -to- C/G/T转换【3】;缺失转换,如用于检测假尿嘧啶的Ψ-to-deletion转换【4,5】。结合高通量测序技术,BC 方法能够识别全基因组/全转录组的修饰位点,精度优于基于免疫沉淀的方法。
然而,多种多样的 BC 方法使数据分析面临着前所未有的挑战。目前,尚无生物信息学工具能够全面处理多样化的 BC 数据。数据比对是其中的关键难题,现有的主要策略包括“突变率策略”和“转换敏感策略”。前者将碱基转换视为错配而产生比对罚分,这会导致reads匹配到错误位置,或被错误丢弃;后者虽然在理论上更加合理,但现有工具尚不支持多路转换和缺失转换等复杂 BC 数据的处理。
为应对这一挑战,同济大学史偈君课题组与北京大学刘君课题组合作,基于核酸序列的位掩码设计和位数运算,开发了BASAL(BAse-conversion Sequencing ALigner)这一新型比对工具,能够准确处理转换碱基的比对罚分,并支持目前所有BC数据的分析(图1)。该成果于近日发表于Nucleic Acids Research,题为BASAL: a universal mapping algorithm for nucleotide base-conversion sequencing【6】。
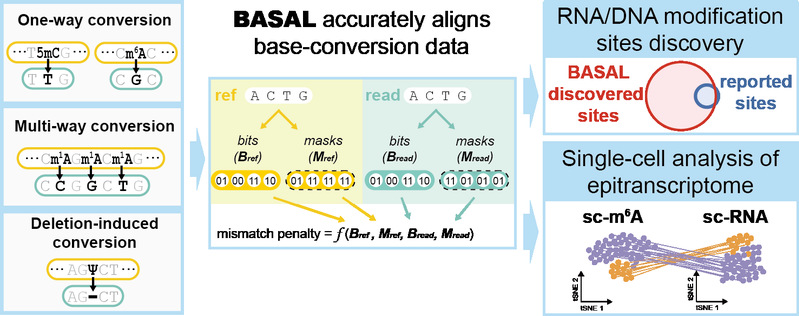
图1、成果概述
BASAL不仅能准确识别已知的修饰位点,还发现了大量新修饰位点(图2)。特别是对于检测RNA假尿嘧啶修饰(Ψ)的诱导缺失转化数据,BASAL比已有工具能发现更多的Ψ位点。通过比较BASAL新发现位点和已知位点的基序,发现 BASAL在识别连续尿嘧啶序列环境中的Ψ方面具有独特的能力,这些位点已被证实与特定生物学功能密切相关【5】。并且,BASAL新发现的Ψ位点也得到了质谱数据和qPCR实验数据的交叉验证,进一步证实了BASAL结果的可靠性。除bulk sequencing数据外,BASAL还改进了单细胞m6A数据的分析,发现了被前人忽视的细胞亚群和分化轨迹,凸显了其在解读单细胞表观转录组学数据方面的巨大潜力。
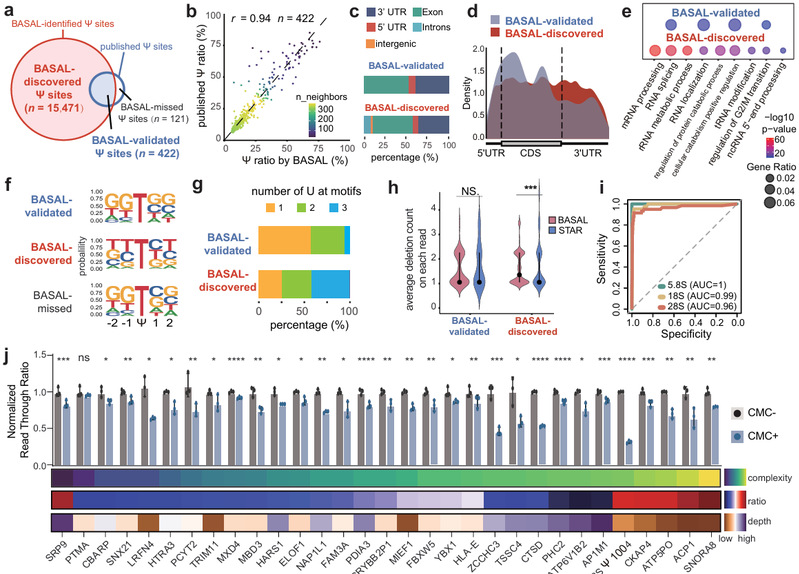
图2、BASAL识别大量未被已有工具发现的RNA假尿嘧啶(Ψ)位点
总之,BASAL 是首个RNA和DNA修饰数据的通用比对算法,能够支持所有碱基转化测序数据的准确分析。由于能正确处理转换碱基的罚分,BASAL显著提高了测序数据的利用率和分析质量,不仅能发现更多可靠的RNA修饰位点,还能准确分析单细胞m6A数据,揭示与生物功能相关的细胞亚群和进化方向,将有助于推动表观基因组学/表观转录组学的突破性发现。
同济大学生命科学与技术学院的史偈君研究员、北京大学生命科学学院的刘君研究员为本文共同通讯作者,史偈君课题组的三年级博士生徐默萍、王淼和刘君课题组的博士生刘潇阳为本文共同第一作者。同济大学高亚威教授、史偈君课题组的二年级研究生罗婷婷也在本工作中有重要贡献。另外,特别感谢北京脑科学与类脑研究中心Magdalena J. Koziol研究员及其博士生冯爽爽分享的单细胞m6A数据。本工作得到了光合基金与国家自然科学基金的资助。
原文链接:https://doi.org/10.1093/nar/gkae1201
BASAL工具发布链接:https://github.com/JiejunShi/BASAL
参考文献
1. Xiao,Y.-L., Liu,S., Ge,R., Wu,Y., He,C., Chen,M. and Tang,W. (2023) Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination. Nat Biotechnol, 10.1038/s41587-022-01587-6.
2. Liu,C., Sun,H., Yi,Y., Shen,W., Li,K., Xiao,Y., Li,F., Li,Y., Hou,Y., Lu,B., et al. (2022) Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat Biotechnol, 10.1038/s41587-022-01487-9.
3. Zhou,H., Rauch,S., Dai,Q., Cui,X., Zhang,Z., Nachtergaele,S., Sepich,C., He,C. and Dickinson,B.C. (2019) Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat Methods, 16, 1281–1288.
4. Dai,Q., Zhang,L.-S., Sun,H.-L., Pajdzik,K., Yang,L., Ye,C., Ju,C.-W., Liu,S., Wang,Y., Zheng,Z., et al. (2022) Quantitative sequencing using BID-seq uncovers abundant pseudouridines in mammalian mRNA at base resolution. Nat Biotechnol, 10.1038/s41587-022-01505-w.
5. Zhang,M., Jiang,Z., Ma,Y., Liu,W., Zhuang,Y., Lu,B., Li,K., Peng,J. and Yi,C. (2023) Quantitative profiling of pseudouridylation landscape in the human transcriptome. Nat Chem Biol, 10.1038/s41589-023-01304-7.
6. Xu,M., Liu,X., Wang,M., Luo,T., Gao,Y., Liu,J., Shi,J. (2024) BASAL: a universal mapping algorithm for nucleotide base-conversion sequencing. Nucleic Acids Res, 10.1093/nar/gkae1201.
Copyright© 2011-2015 生命科学与技术学院, All rights reserved
地址:上海市四平路1239号 电话:021-65981041 传真:65981041
