
科研进展
Nucleic Acids Research|王晨飞课题组开发基于单细胞表观数据构建基因调控网络方法SCRIP
发布时间:2022-09-27
基因表达调控是许多生物学过程的基础,而转录因子结合对于基因表达具有重要的调控作用。近年来,高通量单细胞测序技术的发展,使得研究者们能够从基因的转录组[1]、表观组[2]以及3D结构[3]等不同纬度研究基因表达调控关系,但受技术限制,单细胞转录因子结合与调控的研究一直进展缓慢。单细胞染色质开放测序技术(Single-Cell Assay for Transposase Accessible Chromatin with high-throughput Sequencing, scATAC-seq)可以获得单个细胞水平上的全基因组范围内的染色质开放区域,能够反映单个细胞内部的基因表达调控潜能[4]。然而,这种技术无法获得具体转录因子对单个细胞内基因的调控关系。此外,由于一个细胞中DNA含量较少和受限于scATAC-seq技术的捕获效率,数据具有极大的噪音和稀疏性。这些都为基于scATAC-seq技术研究单细胞水平的基因调控关系带来了巨大挑战。
2022年9月26日,同济大学王晨飞课题组在Nucleic Acids Research杂志上在线发表文章Single-cell gene regulation network inference by large-scale data integration,开发了基于scATAC-seq数据构建基因调控网络的工具SCRIP,基于数据检索和调控潜能模型算法,整合scATAC-seq数据及海量公共数据集来源的转录因子结合数据,衡量单细胞尺度转录因子富集情况,并构建单细胞基因表达调控网络。

SCRIP (Single-Cell gene Regulation network Inference using ChIP-seq and motif) 整合4000余套高质量的人类和小鼠ChIP-seq数据提供的转录调控因子作用的位置信息以及motif序列信息,构建了包含1252个人类和997个小鼠转录因子在不同细胞类型中的结合位点参考集。通过与scATAC-seq数据提供的单细胞水平的染色质开放位置信息进行比对,推断单个细胞内的转录调控因子活性。并利用调控潜能模型[5]推断活跃的转录调控因子下游的靶向基因,从而构建单细胞尺度的基因调控网络。此外,SCRIP还能基于预测的单细胞转录因子调控活性进行细胞聚类,谱系追踪,器官发育以及疾病特异的转录因子调控推断(图1)。
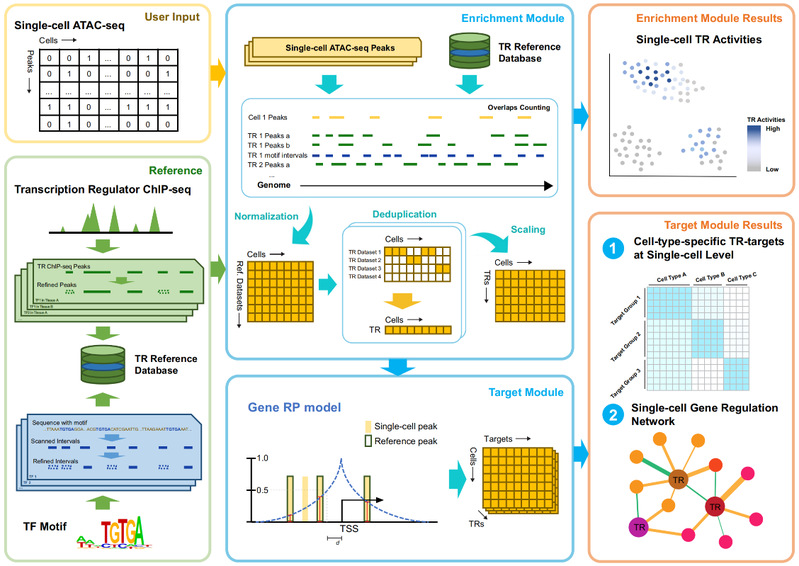
图1 SCRIP 工作流程示意图
为了论证SCRIP在推断转录调控因子活性上的准确性,文章从外周血单核细胞的scATAC-seq数据中推断单个细胞内转录调控因子活性,SCRIP能够鉴定细胞类型特异性的转录调控因子(图2A)。随后,文章比较了这套数据转录组测序提供的细胞注释与SCRIP推断的聚类结果的一致性,与其他已发表的scATAC-seq聚类工具相比,SCRIP基于转录调控因子活性的聚类展现了更好的聚类效果(图2B)。与基于motif序列信息来推断转录调控因子活性的方法[6]相比,SCRIP推断出来的活跃的转录调控因子与其基因的表达水平具有更高的相关性(图2C)。此外,与基于motif序列推断转录调控因子活性的方法相比,SCRIP还能够区分具有相似motif序列却在不同细胞类型中发挥不同功能的转录
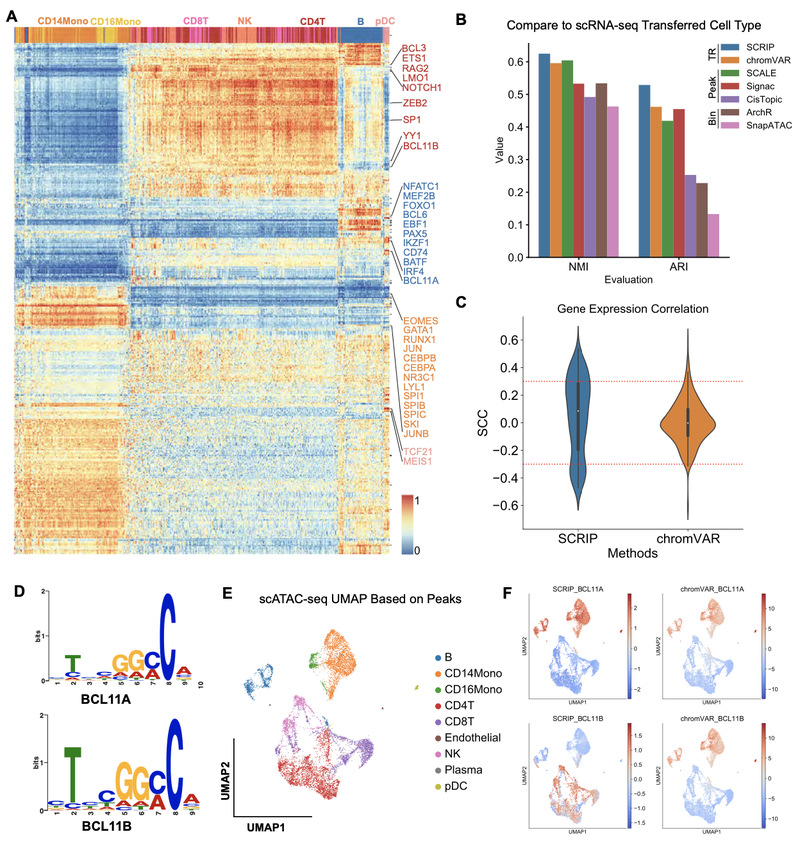
调控因子(图2D-F)。
图2 SCRIP 准确性比较
为了进一步论证SCRIP可被广泛用于不同组织以及疾病状态下的scATAC-seq数据的分析,文章将其应用于人造血干细胞、新生组织器官和基底细胞癌肿瘤微环境的scATAC-seq测序数据中。在人造血干细胞数据中,SCRIP基于转录调控因子活性能够推断细胞类型的分化轨迹,鉴定调控细胞分化的关键转录因子(图3 A, B)。应用于人类新生组织器官数据,在多个器官中均鉴定了相应细胞类型的高活性转录调控因子,基于转录因子活性,也成功地对细胞进行了分群(图3 C, D)。针对于肿瘤免疫微环境,SCRIP首先沿着细胞伪时间轨迹鉴定了T细胞不同时期的关键转录调控因子,并且预测了转录因子JUNB在不同T细胞亚型中可能调控不同的靶基因,进一步可能调控T细胞的状态和表型(图3E, F)。
总的来说,SCRIP利用大量公共的ChIP-seq数据提供的转录因子作用位置信息与motif序列信息作为参考,整合scATAC-seq数据,计算单细胞尺度上的转录调控因子活性,并可以用于多个系统中转录调控因子活性计算以及单细胞尺度上的基因调控网络的构建。
同济大学生命科学与技术学院王晨飞研究员为该论文通讯作者,同济大学生命科学与技术学院博士研究生董鑫、唐珂为文章的共同第一作者。该项工作得到了国家自然科学基金委及上海市科委等项目的重要支持。
同济大学王晨飞课题组聚焦单细胞及空间多组学数据机器学习方法开发,并将其应用于解决肿瘤免疫微环境、胚胎发育过程中表观修饰及空间信息的异质性,探索其对细胞状态改变及命运决定的调控机制。课题组长期招收计算生物学、单细胞多组学方向的研究生及博士后,欢迎大家加入,联系邮箱:08chenfeiwang@tongji.edu.cn。
参考文献:
1. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628.
2. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al. . Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330.
3. Ramani V., Deng X., Qiu R., Gunderson K.L., Steemers F.J., Disteche C.M., Noble W.S., Duan Z., Shendure J. Massively multiplex single-cell Hi-C. Nat. Methods. 2017; 14:263–266.
4. Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015; 523:486–490.
5. Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y. et al. . Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020; 21:198.
6. Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017; 14:975–978.
Copyright© 2011-2015 生命科学与技术学院, All rights reserved
地址:上海市四平路1239号 电话:021-65981041 传真:65981041
