
科研进展
PNAS | 毛志勇/魏珂团队揭示儿童早衰症小鼠DNA修复缺陷导致心肌萎缩发生的分子机制并提供潜在干预对策
发布时间:2023-11-22
儿童早衰症(Hutchinson-Gilford progeria syndrome, HGPS)是一种罕见的衰老加速疾病,患者平均寿命约14.5年,心血管疾病是其致死的最主要原因[1, 2]。然而,儿童早衰症患者心脏的病变机制还不明确,当前也缺乏有效的针对性治疗手段。许多研究表明:DNA损伤积累与衰老发生关系密切。与其他早衰症类似,HGPS患者细胞存在明显的基因组不稳定特征[3]。然而,HGPS相关DNA损伤的来源为何?其是否及如何导致了心脏病变的产生?上述问题仍未得到很好地回答。
2023年11月15日,同济大学附属第一妇婴保健院、生命科学与技术学院及附属东方医院毛志勇/魏珂团队在PNAS杂志发表了题为“Impaired end joining induces cardiac atrophy in a Hutchinson-Gilford progeria mouse model”的研究论文(图1)。在该工作中,研究团队成员发现:HGPS小鼠心脏及心肌细胞变小,心脏功能显著下降,呈现心肌萎缩的表型。RNA-seq数据分析发现HGPS小鼠心脏发育、细胞尺寸等通路基因的表达显著改变。此外,DNA损伤应答相关基因的表达水平在HGPS小鼠心脏中亦存在显著变化。这驱使团队成员围绕HGPS小鼠心脏的DNA修复开展研究。

团队成员在前期工作中开发了多种DNA修复的定量报告系统[4-7]。然而,由于成年小鼠心肌细胞分离、体外培养及转染存在较大难度,如何在体内利用报告系统定量研究心脏中的DNA修复成为亟待解决的技术难点。为此,团队成员基于前述研究基础,建立了一种新的DNA修复体内报告模型,实现了心肌中DNA修复的高效在体检测(图2)。利用这一模型,研究人员发现:HGPS小鼠模型的心肌细胞中存在大量DNA双链断裂积累及基因组不稳定现象,这来源于双链断裂修复通路中非同源末端连接(NHEJ)通路修复能力的显著降低。进一步机制研究表明:HGPS的致病基因Lmna的突变使其编码的突变体Lamin A蛋白(称之为早衰素,Progerin)与损伤位点的标志物γH2AX互作减弱,干扰其招募至损伤位点,也影响了下游NHEJ因子的进一步募集。
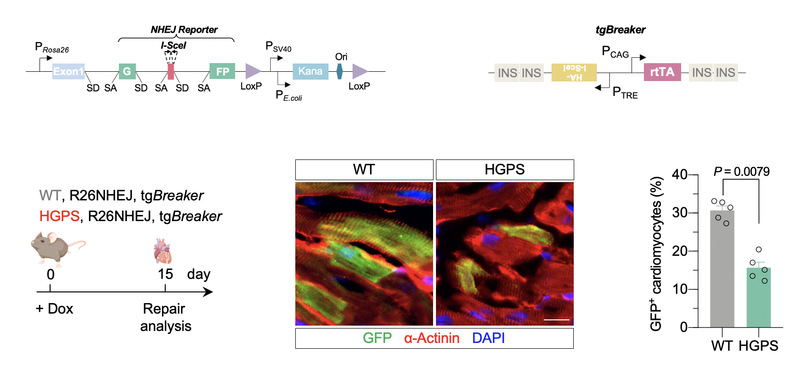
图2: 儿童早衰症心肌细胞NHEJ修复能力显著下降
(绿色荧光示NHEJ事件,红色荧光示心肌细胞标记物α-Actinin)
DNA损伤的积累是否直接导致心肌萎缩发生?团队成员发现:HGPS小鼠心脏中出现了损伤应答上游激酶CHK2的表达提升与激活。而有趣的是,被认为可拮抗心肌肥大发生的蛋白AMPKα亦在HGPS小鼠心脏中呈现高度磷酸化的激活状态。由此,研究人员猜测:AMPKα的持续性激活可能破坏了心脏稳态,最终介导了心肌萎缩的发生。然而:CHK2是否引起了AMPKα的激活?进一步的机制研究发现:CHK2以一种不依赖于经典酶活的方式,促进了AMPKα与其上游激酶LKB1的相互作用,进而导致AMPKα磷酸化的增加及下游转录因子FOXO3A的激活。FOXO3A的激活进一步促进了两种肌肉萎缩相关基因Fbxo32和Trim63的转录水平提升,最终介导了心肌萎缩的发生。而该发现亦被公共RNA-seq数据所验证。
为探寻可能的干预手段,团队成员首先利用AMPK的酶活抑制剂处理心肌细胞,发现其可以显著增大心肌细胞尺寸。AMPK的酶活抑制剂的体内给药亦缓解了HGPS小鼠的心肌萎缩表型。然而,AMPK的酶活抑制剂可能干扰机体的能量稳态,这一定程度上限制了它的临床转化。因此,研究团队成员进而探索异丙肾上腺素(ISO)这一已被临床批准使用的药物在治疗HGPS相关心肌萎缩方面的潜力。ISO在实验室中被广泛用于诱导动物模型的病理性心肌肥大与心力衰竭[8]。然而,对HGPS小鼠进行为期两周的间断性ISO给药可促进HGPS小鼠心脏的肥大性生长,使原本呈萎缩表型的心脏回复到与野生型小鼠相当的尺寸。值得注意的是,ISO处理拯救了HGPS小鼠的心功能衰退,并显著延长了HGPS小鼠的存活时间(~20%)(图3)。
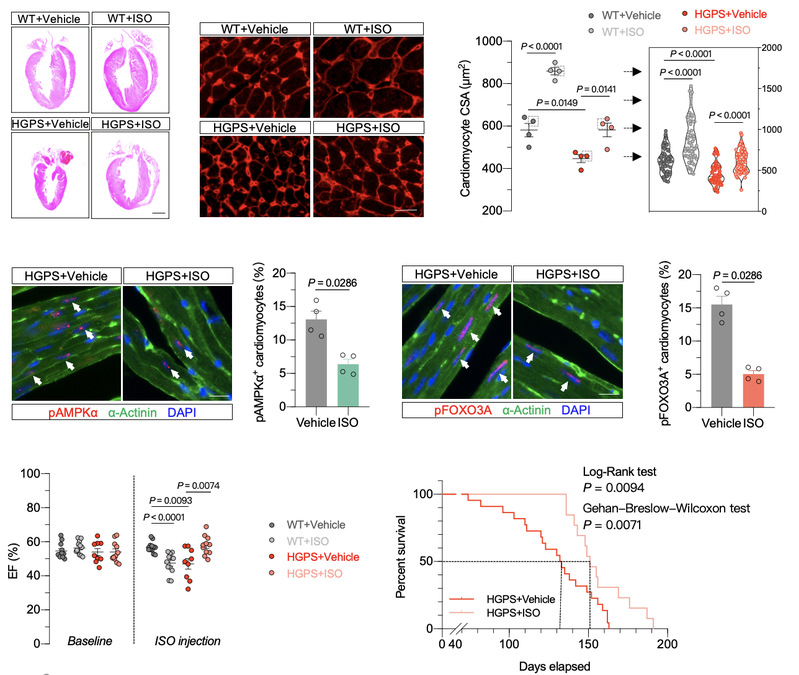
图3: ISO处理拯救儿童早衰症小鼠心肌萎缩表型并延长小鼠寿命
总之,研究团队的该项工作揭示了HGPS小鼠心肌中DNA双链断裂修复的异常介导器官功能退化的分子机制,为靶向干预心肌萎缩提供了潜在靶点通路和极具临床价值的候选方案(图4)。
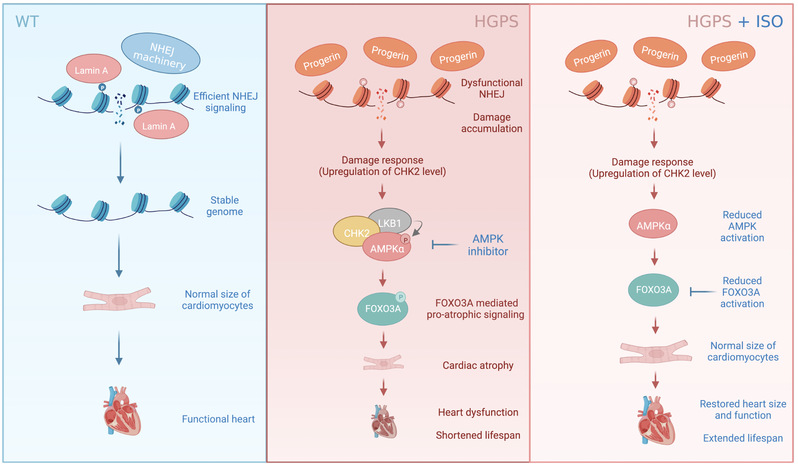
图4: DNA损伤修复下调诱导HGPS小鼠心肌萎缩的机制与干预模式图
同济大学附属第一妇婴保健院/生命科学与技术学院毛志勇教授、同济大学生命科学与技术学院/附属东方医院魏珂教授为该论文的共同通讯作者。同济大学生命科学与技术学院助理教授陈御及博士研究生黄诗琪为该论文的共同第一作者。参与此项研究工作的还有崔震,孙小翔博士,唐岩松博士,博士研究生张红杰、陈芷茜、姜睿,助理研究员张伟娜,硕士研究生李雪及蒋颖副教授。本工作还得到了同济大学陈嘉瑜教授和深圳大学刘宝华教授的大力支持。该项工作得到了科技部重点研发计划、国家自然科学基金、上海市科委项目的资助。
参考文献:
1.De Sandre-Giovannoli, A., et al., Lamin a truncation in Hutchinson-Gilford progeria. Science, 2003. 300(5628): p. 2055.
2.Hennekam, R.C., Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A, 2006. 140(23): p. 2603-24.
3.Liu, B.H., et al., Genomic instability in laminopathy-based premature aging. Nature Medicine, 2005. 11(7): p. 780-785.
4.Chen, Y., et al., A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Research, 2019. 47(16): p. 8563-8580.
5.Chen, Y., et al., IDDoR: A novel reporter mouse system for simultaneous and quantitative in vivo analysis of both DNA double-strand break repair pathways. Protein & Cell, 2022: p. pwac001.
6.Zhang, W., et al., A high-throughput small molecule screen identifies farrerol as a potentiator of CRISPR/Cas9-mediated genome editing. eLife, 2020. 9.
7.Wang, C., et al., Rational combination therapy for hepatocellular carcinoma with PARP1 and DNA-PK inhibitors. Proc Natl Acad Sci U S A, 2020. 117(42): p. 26356-26365.
8.Sniecinski, R.M., S. Wright, and J.H. Levy, Chapter 3 - Cardiovascular Pharmacology, in Cardiothoracic Critical Care, D. Sidebotham, et al., Editors. 2007, Butterworth-Heinemann: Philadelphia. p. 33-52.
Copyright© 2011-2015 生命科学与技术学院, All rights reserved
地址:上海市四平路1239号 电话:021-65981041 传真:65981041
